
Pipeline DM1
Delivering on the RNA Revolution
We are advancing and expanding our innovative AOC pipeline to offer treatment options for patients and their families across a wide range of therapeutic areas. We have three AOC programs for three distinct rare diseases in clinical development from our muscle disease franchise: myotonic dystrophy type 1 (DM1), facioscapulohumeral muscular dystrophy (FSHD), and Duchenne Muscular Dystrophy (DMD). Our pipeline also includes advancing AOCs to address additional DMD and rare neuromuscular programs.
Myotonic Dystrophy Type 1
Myotonic dystrophy type 1 (DM1) is an underrecognized, progressive and often fatal neuromuscular disease with no approved therapies. DM1 is estimated to affect an estimated 80,000 in the United States and Europe. DM1 primarily affects skeletal and cardiac muscle, however people can suffer a range of symptoms including myotonia and muscle weakness, respiratory and cardiac problems, severe gastrointestinal complications, and cognitive and behavioral impairment—resulting in significant decline in quality of life and large caregiver burden. The disease is highly variable with respect to disease severity, presentation, and age of onset.
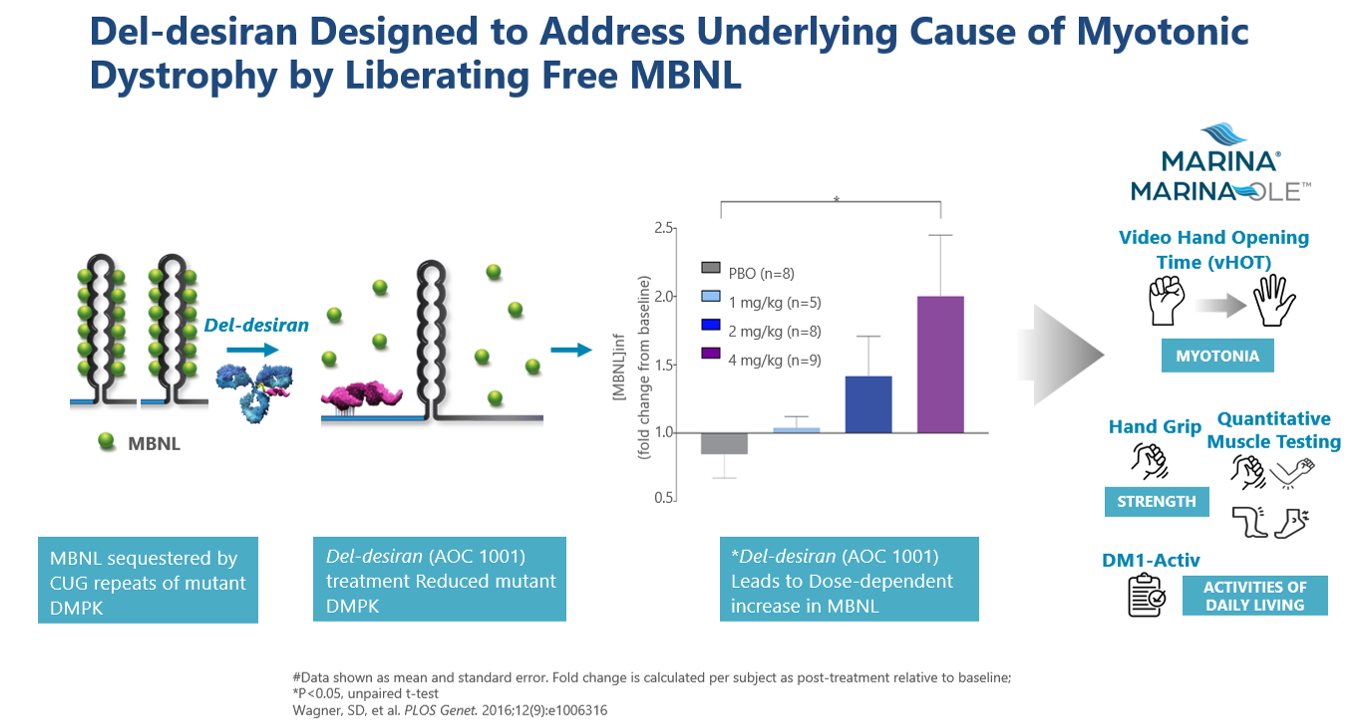
DM1 is caused by an increase in the number of CUG triplet repeats found in the myotonic dystrophy protein kinase (DMPK) gene. In a healthy individual the number of repeats is approximately 35 but in someone with DM1 there can be thousands. When there are too many CUG repeats, large hairpin loops form trapping DMPK mRNA in the nucleus, effectively sequestering muscleblind-like protein (MBNL) and reducing its activity. Specifically, mutated DMPK mRNA binds to MBNL, a critical CUG-binding protein, forming nuclear foci and preventing MBNL from performing its normal function of processing the mRNAs for many other genes. As a result, multiple mRNAs that encode key proteins are misprocessed, resulting in atypical proteins that ultimately are the cause of DM1. Our therapeutic approach is to reduce DMPK and CUG levels thereby reducing nuclear foci and relieving the sequestration of MBNL so that MBNL can perform its normal function.

Del-desiran is designed to address the root cause of DM1 by reducing levels of a disease-related mRNA called DMPK in skeletal, cardiac, and smooth muscle. Del-desiran consists of a proprietary monoclonal antibody (mAb) that binds to the transferrin receptor 1 (TfR1) conjugated with a small interfering RNA (siRNA) targeting DMPK mRNA. This allows del-desiran to address the underlying cause of disease by reducing DMPK mRNA, releasing MBNL as shown in MARINA® participants where del-desiran treatment led to dose-dependent increases in inferred MBNL and, potentially alleviating the spectrum of symptoms that people with DM1 experience.
Delpacibart etedesiran, or del-desiran (formerly AOC 1001)
Del-desiran, Avidity’s investigational product candidate for DM1 utilizing its AOC platform is being studied in the Phase 3 global HARBOR™ trial in adolescents and adults living with the disease. Del-desiran is also being studied in the ongoing MARINA-OLE™ trial with all of the participants who completed the Phase 1/2 MARINA® trial. The U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) granted Orphan designation for del-desiran and the FDA has granted del-desiran Fast Track designation and Breakthrough Therapy designation.
Data Update at 2024 Muscular Dystrophy Association (MDA) Clinical & Scientific Conference
In March 2024, Avidity announced new positive long-term del-desiran data from the MARINA-OLE™ trial showing reversal of disease progression in people living with DM1 across multiple endpoints including vHOT, muscle strength and activities of daily living compared to a matched END-DM1 natural history study population over one year. These endpoints are the same key endpoints that will be used in the global Phase 3 HARBOR™ trial for people living with DM1. The primary endpoint in the Phase 3 HARBOR trial is video hand opening time (vHOT), a measurement of hand function and myotonia. Key secondary endpoints include muscle strength as measured by hand grip strength and quantitative muscle testing (QMT) total score, and activities of daily living as measured by DM1-Activ, a patient reported outcome (PRO).
The global Phase 3 HARBOR™ trial is a randomized, placebo-controlled, double-blind pivotal study designed to evaluate del-desiran in approximately 150 people (age 16 and older) living with DM1. The trial will be conducted at approximately 40 sites globally. Patients will be administered either del-desiran or placebo (1:1) every eight weeks. The trial is designed to assess del-desiran’s impact on multiple key aspects of DM1 including myotonia, muscle strength and activities of daily living. All study participants, regardless if they receive active treatment or placebo, will have the option to enroll into an open-label extension trial. For more information about the HARBOR trial, visit the HARBOR study website or visit https://www.clinicaltrials.gov and search for NCT06411288.
MARINA-OLE™ is an open-label, multi-center trial designed to evaluate the long-term safety and tolerability of del-desiran in participants with myotonic dystrophy type 1 (DM1) who were previously enrolled in the MARINA® Phase 1/2 trial. This trial continues to evaluate the safety, tolerability, PK, PD, and efficacy of del-desiran in participants enrolled in the randomized, placebo-controlled, Phase 1/2 MARINA clinical trial. All participants previously enrolled in the MARINA study rolled-over into the MARINA-OLE study and continue to receive del-desiran regardless of whether they received treatment or placebo in the MARINA study. For more information on this study click here or visit https://www.clinicaltrials.gov and search for NCT05479981.
The MARINA® trial was a randomized, double-blind, placebo-controlled, Phase 1/2 clinical trial that enrolled 38 adults with DM1. The primary objective of this study was to evaluate the safety and tolerability of single and multiple ascending doses of del-desiran administered intravenously. The MARINA trial assessed the activity of del-desiran across key biomarkers, including spliceopathy, an important biomarker for DM1, and knockdown of DMPK mRNA. Though the Phase 1/2 trial was not powered to assess functional benefit, it explored the clinical activity of del-desiran in multiple measures of muscle function including myotonia, muscle strength, measures of mobility as well as patient reported outcomes and quality of life measures. Patients had the option to enroll in MARINA-OLE™, an open label extension study, at the end of the post-treatment period. For more information on this study click here or visit https://www.clinicaltrials.gov and search for NCT05027269.